1. Molecular cloning and protein work
 Our key research focus centers on the function of distinct molecular players of the endocannabinoid system and other lipid signaling pathways in the nervous system. To address our research questions, we have generated an extensive library of genetic constructs, allowing us to perform correlative experiments (e.g. using HA-, SEP-, AcGFP- or mCherry-tagged proteins) and perturbation experiments (e.g. using gain-of-function or loss-of-function models) against our genes of interest. Therefore, the laboratory is well equipped for all standard molecular neurobiology techniques, which are critical for all of our current projects. We occasionally receive DNA constructs from colleagues and commercial sources, but we also regularly generate plasmids in the lab. We achieve this by firstly purifying total RNA from brain tissue, reverse transcribing it into cDNA and then by using conventional PCR we can obtain enough DNA for plasmid generation. We have two thermocyclers (MJ Research PTC-200 and BIO-RAD C1000). The latter of which has a temperature gradient function, allowing rapid and accurate reaction optimization. Also, the lab maintains several genetically-modified mouse strains (>10 lines at the moment), therefore the thermocyclers are regularly used for genotyping reactions.
Our key research focus centers on the function of distinct molecular players of the endocannabinoid system and other lipid signaling pathways in the nervous system. To address our research questions, we have generated an extensive library of genetic constructs, allowing us to perform correlative experiments (e.g. using HA-, SEP-, AcGFP- or mCherry-tagged proteins) and perturbation experiments (e.g. using gain-of-function or loss-of-function models) against our genes of interest. Therefore, the laboratory is well equipped for all standard molecular neurobiology techniques, which are critical for all of our current projects. We occasionally receive DNA constructs from colleagues and commercial sources, but we also regularly generate plasmids in the lab. We achieve this by firstly purifying total RNA from brain tissue, reverse transcribing it into cDNA and then by using conventional PCR we can obtain enough DNA for plasmid generation. We have two thermocyclers (MJ Research PTC-200 and BIO-RAD C1000). The latter of which has a temperature gradient function, allowing rapid and accurate reaction optimization. Also, the lab maintains several genetically-modified mouse strains (>10 lines at the moment), therefore the thermocyclers are regularly used for genotyping reactions.
We exploit most conventional bacterial systems for initial cloning and the resulting plasmids are analyzed on agarose gels by restriction enzyme digestions or by polymerase chain reactions and are always sequenced before use. We also utilize the E. coli heterologous protein expression system to purify GST-tagged epitopes for antibody generation. Expressed proteins can be analyzed via conventional SDS polyacrylamide gel electrophoresis. The GST tags are cut from the epitope using thrombin, and injected into rabbits or KO mice to generate the desired antisera (this latter step is carried out in collaboration with Prof. Imre Kacskovics at the Department of Immunology, Eötvös Loránd University, Budapest and Immunogenes AG). Following immunization, the affinity-purified antibodies are initially analyzed on a Western blot, then verified by immunohistochemistry on brain sections derived from wild-type and knock-out animals. All of our published constructs and antibodies are freely available for the life science community upon request.
2. Quantitative real-time PCR
Besides conve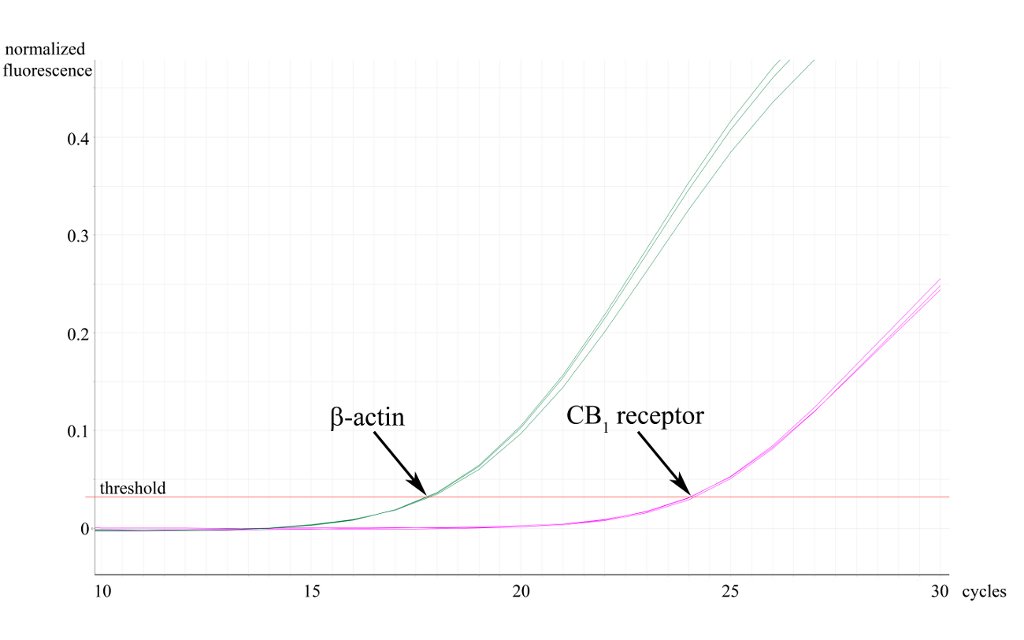 ntional end-point PCR, we also exploit qPCR to investigate the expression level of our genes of interest in distinct tissues or brain regions. In addition, real time PCR can also be a useful way of validating conventional genotyping experiments. To analyse mRNA expression levels, we first extract total RNA from distinct brain regions (and other tissues), then reverse transcribe to cDNA, from which our genes of interest are amplified using the real time DyNAmo SYBRGreen PCR technology (Finnzymes). For genotyping, we use genomic DNA samples purified from mouse tails. qPCR reactions are performed on a Rotor Gene 3000 (Corbett Research, now Qiagen). Data is collected using the latest Rotor-Gene 6 software and we follow the principles of the Pfaffl-method for quantitative analysis.
ntional end-point PCR, we also exploit qPCR to investigate the expression level of our genes of interest in distinct tissues or brain regions. In addition, real time PCR can also be a useful way of validating conventional genotyping experiments. To analyse mRNA expression levels, we first extract total RNA from distinct brain regions (and other tissues), then reverse transcribe to cDNA, from which our genes of interest are amplified using the real time DyNAmo SYBRGreen PCR technology (Finnzymes). For genotyping, we use genomic DNA samples purified from mouse tails. qPCR reactions are performed on a Rotor Gene 3000 (Corbett Research, now Qiagen). Data is collected using the latest Rotor-Gene 6 software and we follow the principles of the Pfaffl-method for quantitative analysis.
3. Histology
For the majority of neuroanatomy experiments evaluated by light microscopy (e.g. non-radioactive free-floating in situ hybridization, immunoperoxidase and immunofluorescence stainings), we use 30-60 µm-thick brain sections. After perfusion, sections are cut from the fixed tissue blocks with a Leica VT1000 S Vibrating Blade Microtome. The variable frequency allows the VT1000 S to perform well, even under situations of inconsistent tissue quality. For multiple-fluorescence in situ hybridization experiments, tissue is first embedded in OCT and frozen. Then, 10 µm-thick sections are cut with a Microm HM 550 cryostat and mounted onto glass slides. Semi-thin and ultra-thin sections for light and electron microscopy respectively, are prepared using a Leica EM UC6 Ultramicrotome .
4. In situ hybridization
In situ hybridization is routinely used to initially identify the cells that express our gene of interest. First, we generate digoxigenin-labelled 1000-2000 bp-long riboprobes that are complementary (antisense probe) or identical (sense probe, used as a method control) to the mRNA of interest using in vitro transcription. Subsequently, these riboprobes are hybridized to mRNA molecules in brain sections using a free-floating approach and stringent conditions (65 oC; 5xSSC, 50% formamide). After extensive washing, digoxigenin-labelled riboprobe-mRNA duplexes are identified by an anti-digoxigenin antibody that is coupled to an alkaline phosphatase enzyme. Finally, the localization of the target mRNA is visualized by adding the nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate substrates of the alkaline phosphatase, which form a blue precipitate inside the cells as a result of enzymatic activity.
5. Immunohistochemistry
 To uncover the precise regional, cellular and subcellular localization of distinct proteins, we use three complementary methods of immunohistochemistry.
To uncover the precise regional, cellular and subcellular localization of distinct proteins, we use three complementary methods of immunohistochemistry.
We use the immunoperoxidase approach primarily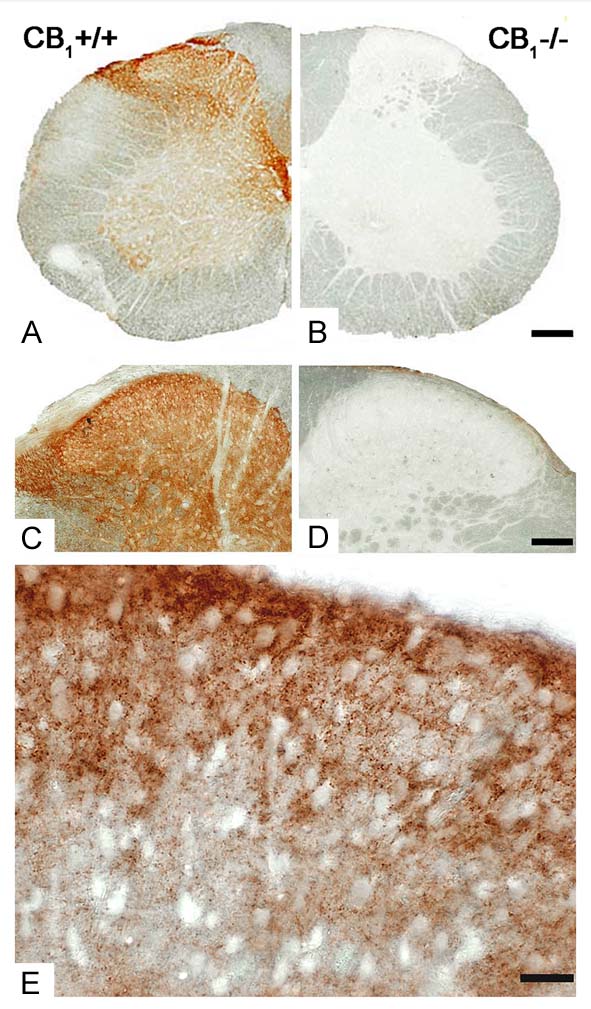 for its sensitivity and stability. The chromogen of choice is 3,3′-diaminobenzidine (DAB). This approach has been optimized to reveal the regional distribution of a given protein at the light microscopic level, but can also be exploited to expose the subcellular distribution at the electron microscopic level. For light microscopic analysis, specific regions of the CNS are analyzed and photographed using a Nikon Eclipse 80i upright microscope with high quality objectives (e.g. Plan Apo VC DIC 60x oil with NA1.40) and a Nikon DS-Fi1 digital color camera.
for its sensitivity and stability. The chromogen of choice is 3,3′-diaminobenzidine (DAB). This approach has been optimized to reveal the regional distribution of a given protein at the light microscopic level, but can also be exploited to expose the subcellular distribution at the electron microscopic level. For light microscopic analysis, specific regions of the CNS are analyzed and photographed using a Nikon Eclipse 80i upright microscope with high quality objectives (e.g. Plan Apo VC DIC 60x oil with NA1.40) and a Nikon DS-Fi1 digital color camera.
Immunofluorescence is regularly exploited in the lab for its expedient ability to reveal the cellular expression pattern of more than one protein in the same sample. Our Nikon Eclipse 80i upright microscope is equipped with an epi-fluorescent attachment, containing FITC, Texas Red, DAPI and Cy5 filter cubes, plus a Nikon Super High Pressure Mercury lamp. However, most of the detailed analysis is performed on the Nikon A1R confocal system, which is based on a Nikon Ti-E inverted microscope. It is equipped with 4 lasers providing 405, 458, 488, 514, 561, 641 nm laser lines as well as with a 32-channel spectral detector, greatly enhancing our flexibility in the design of multi-coloured staining experiments. Notably, recent technological advances also offer that multiple targets can be studied at the subcellular level. We are routinely using the STORM super-resolution technology to uncover the subcellular distribution of target proteins with a 20-40 nm resolution.
 Finally, we routinely apply immunogold labeling technology to reveal the precise subcellular position of a given protein at a nanometer scale. Target proteins in fixed brain sections are bound by antibodies conjugated to 0.8 nm gold particles, which are further enhanced using a silver intensification approach. The immunostained sections are then treated with OsO4 and uranyl acetate, dehydrated in an ascending series of ethanol and acetonitrile, and embedded in Durcupan. Areas of interest are re-embedded and re-sectioned. Electron
Finally, we routinely apply immunogold labeling technology to reveal the precise subcellular position of a given protein at a nanometer scale. Target proteins in fixed brain sections are bound by antibodies conjugated to 0.8 nm gold particles, which are further enhanced using a silver intensification approach. The immunostained sections are then treated with OsO4 and uranyl acetate, dehydrated in an ascending series of ethanol and acetonitrile, and embedded in Durcupan. Areas of interest are re-embedded and re-sectioned. Electron microscopic analysis of 20-60 nm thick ultra-thin sections on Formvar-coated single-slot grids is performed using a Hitachi 7100 electron microscope. Electron micrographs are taken at 1,000 – 100,000x magnification.
microscopic analysis of 20-60 nm thick ultra-thin sections on Formvar-coated single-slot grids is performed using a Hitachi 7100 electron microscope. Electron micrographs are taken at 1,000 – 100,000x magnification.
One word of caution; all three immunohistochemistry approaches are critically dependent on the availability of a highly sensitive, but also highly specific antibody which recognizes our target protein in fixed brain tissue. Although most antibodies work well on Western-blots, according to our and several other colleagues’ experiences, the vast majority of published or commercial antibodies (90%) are not specific enough to be used in fixed tissue! Thus, we validate the specificity of a given immunostaining pattern in corresponding knockout tissue. When knockouts are not available, we use two independent antibodies, raised against independent epitopes. The latter approach is not unequivocal, but largely reduces the probability that a given pattern visualized by both antibodies is due to an off-target effect.
6. Immunocytochemistry and live-cell imaging
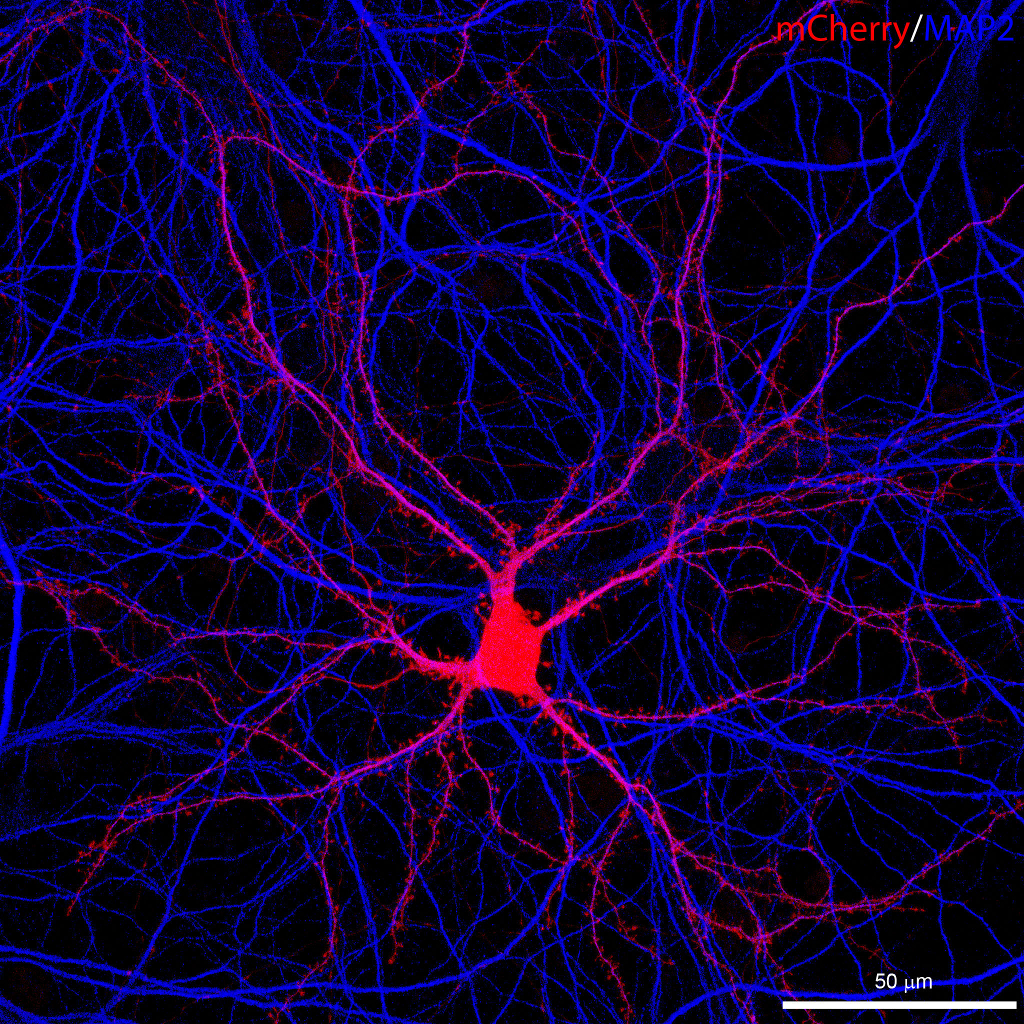 We use several cell culture preparations and express various constructs in these cells to study the operation and function of endocannabinoid-mediated or endo-cannabinoid-related signaling pathways. Frequently, we use primary hippocampal cultures, which are prepared from P1 mice. Cells are dissociated after papain digestion by trituration with fire-polished glass pipettes and plated onto poly-D-lysine coated glass coverslips or on glass bottom petri dishes under an ESCO Labculture laminar box. We maintain the cultures at 37°C and 5% CO2 in a Thermo Forma incubator in Neurobasal A media supplemented with B-27 and 2 mM L-glutamine. Transfections with plasmid DNA are performed using Lipofectamine 2000. Organotypic hippocampal slice cultures are prepared from vibratome slices of P6 mice, and maintained on culture plate inserts in media consisting of 50% MEM, 25% HBSS, 25% NHS, 30 mM glucose, 1 mM Glutamax-I, and penicillin-streptomicin. To transfect the organotypic slices, we use the Helios Gene Gun System from Bio-Rad.
We use several cell culture preparations and express various constructs in these cells to study the operation and function of endocannabinoid-mediated or endo-cannabinoid-related signaling pathways. Frequently, we use primary hippocampal cultures, which are prepared from P1 mice. Cells are dissociated after papain digestion by trituration with fire-polished glass pipettes and plated onto poly-D-lysine coated glass coverslips or on glass bottom petri dishes under an ESCO Labculture laminar box. We maintain the cultures at 37°C and 5% CO2 in a Thermo Forma incubator in Neurobasal A media supplemented with B-27 and 2 mM L-glutamine. Transfections with plasmid DNA are performed using Lipofectamine 2000. Organotypic hippocampal slice cultures are prepared from vibratome slices of P6 mice, and maintained on culture plate inserts in media consisting of 50% MEM, 25% HBSS, 25% NHS, 30 mM glucose, 1 mM Glutamax-I, and penicillin-streptomicin. To transfect the organotypic slices, we use the Helios Gene Gun System from Bio-Rad.
Live specimens expressing fluorescently tagged proteins or fixed specimens stained via immunocytochemistry are imaged with the Nikon A1R confocal system on the Ti-E inverted microscope. To maintain stable conditions for live cell imaging, the microscope is also equipped with a whole-stage temperature and CO2 controller, and a perfusion chamber . Stable long-term imaging is further supported with the NIKON Perfect Focus System. The system also includes a piezo z-stage and a resonant scanner to allow fast frame rates, and a 32-channel spectral detector for unmixing of signals with close emission spectra. Among the objectives a 1.4 NA 60x CFI Plan Apochromat VC oil immersion objective is available for high resolution imaging in a wide spectral range. We utilize the Huygens de-convolution software by SVI for restoration of 3D image stacks. Image analysis is performed with ImageJ, Adobe Photoshop, Huygens and NIS elements software.
7. In utero electroporation
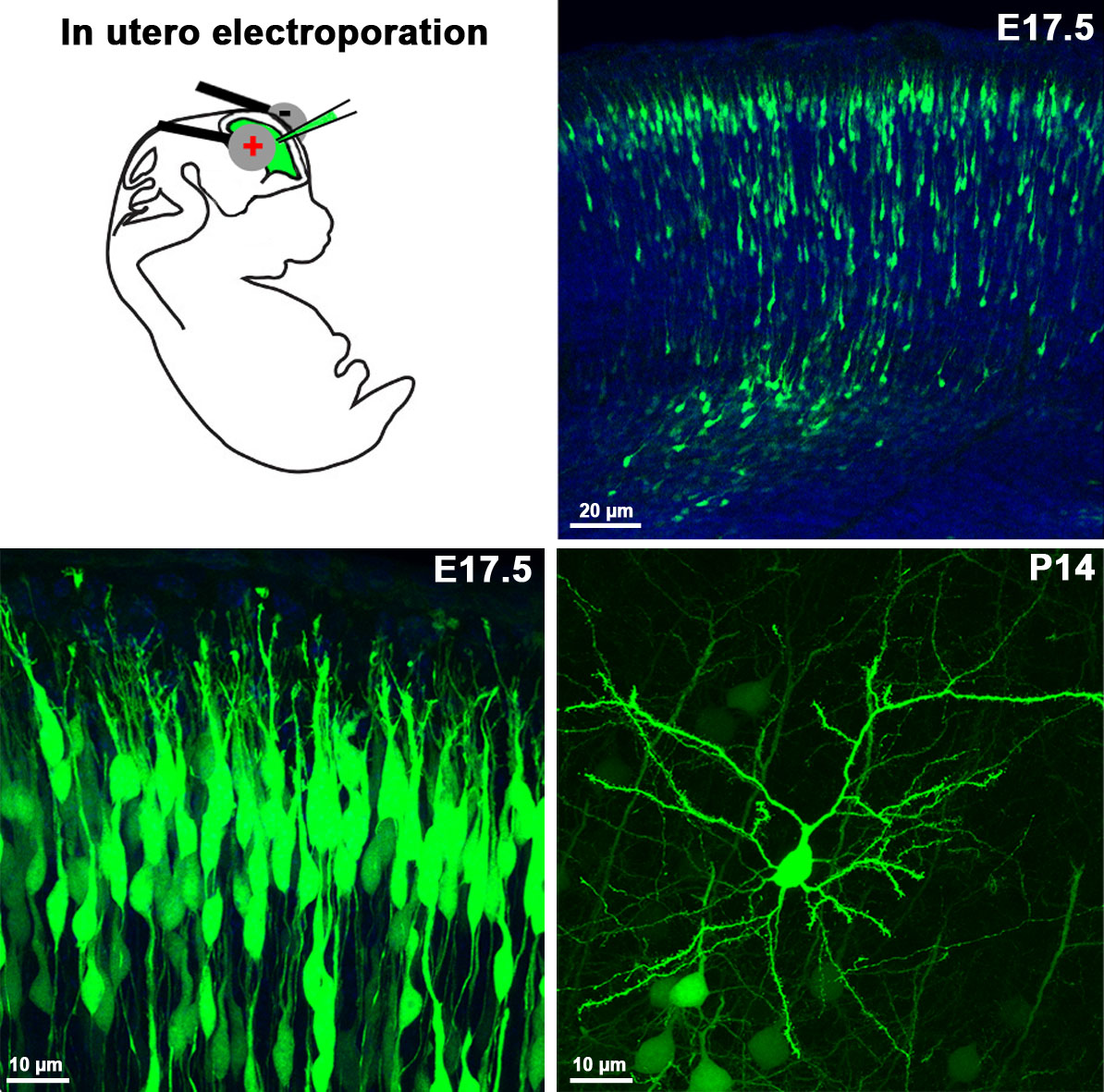 To label specific neuronal populations or to regulate gene expression levels in given cell types (either by overexpression or by siRNA silencing), in utero electroporation is an emerging approach. Its advantages are speed and affordability over the generation of conventional genetically-modified animal models, which are usually slow and very expensive. Moreover, in utero electroporation usually results in animal models which lack functional redundancy, and this approach often circumvents premature lethality. We use plasmids encoding either the mRNA (gain-of-function) or RNAi (loss-of-function) of the gene to be examined, which is injected into the telencephalic ventricle of E12-E16 embryos, followed by a brief electric pulse train of low voltage (30-50V) applied via electrodes held to the head of the embryos. Upon the direct current, the plasmid will pass into the proliferating ventricular zone precursors then enters into the progeny of these cells (neurons or progenitor cells). When a fluorescent protein-containing plasmid is co-injected, or alternatively a GFP- or mCherry-tagged version of the cDNA is injected, one can easily follow the fate of the transfected progenitor and its daughter cells. We also plan to combine this technique with immunhistochemistry and patch-clamp electrophysiology in the near future.
To label specific neuronal populations or to regulate gene expression levels in given cell types (either by overexpression or by siRNA silencing), in utero electroporation is an emerging approach. Its advantages are speed and affordability over the generation of conventional genetically-modified animal models, which are usually slow and very expensive. Moreover, in utero electroporation usually results in animal models which lack functional redundancy, and this approach often circumvents premature lethality. We use plasmids encoding either the mRNA (gain-of-function) or RNAi (loss-of-function) of the gene to be examined, which is injected into the telencephalic ventricle of E12-E16 embryos, followed by a brief electric pulse train of low voltage (30-50V) applied via electrodes held to the head of the embryos. Upon the direct current, the plasmid will pass into the proliferating ventricular zone precursors then enters into the progeny of these cells (neurons or progenitor cells). When a fluorescent protein-containing plasmid is co-injected, or alternatively a GFP- or mCherry-tagged version of the cDNA is injected, one can easily follow the fate of the transfected progenitor and its daughter cells. We also plan to combine this technique with immunhistochemistry and patch-clamp electrophysiology in the near future.
8. Patch-clamp electrophysiology
 To investigate the functional significance of the signaling pathways, which we have uncovered in our molecular biology and anatomy experiments, we perform whole-cell patch-clamp recordings in acute brain slice preparations. We also plan to use organotypic slice cultures in the near future to exploit the advantages of acute genetic perturbations along with pharmacological tools and conventional chronic knockout models. To prepare acute and organotypic slices, we use the latest Leica 1200S Vibratome. Our setup was built on the I-shaped Nikon FN1 upright microscope, and is positioned on a Supertech anti-vibration table. We use a water-immersion 40x CFI Apo objective with NA 0.8 and a working distance of 3.5 mm. Infrared DIC is utilized to enhance contrast in our samples and patching of selected neurons is aided by a Hamamatsu near infrared ½-inch CCD camera (C7500-51). An epifluorescence attachment also allows visualization of cells with genetically-encoded fluorescent markers, which help to identify and target specific cell types required for certain projects. Patch-clamp electrodes are pulled by a Sutter P-1000 horizontal pipette puller. Two Luigs & Newmann micromanipulator systems help us to carry out paired recordings or extracellular stimulation combined with single cell recordings. Whole-cell recordings are made in voltage- or current-clamp mode using an Axoclamp 2B amplifier from Axon Instruments. A second, almost identical setup is also available to us at the Nikon Microscopy Center located within the institute. This setup has a Nikon-C1 confocal attachment and we plan to monitor presynaptic calcium transients and postsynaptic responses simultaneously in our preparations. With the help of these electrophysiological setups, we aim to contribute to the understanding of how distinct endocannabinoid-related signaling pathways regulate certain forms of synaptic plasticity.
To investigate the functional significance of the signaling pathways, which we have uncovered in our molecular biology and anatomy experiments, we perform whole-cell patch-clamp recordings in acute brain slice preparations. We also plan to use organotypic slice cultures in the near future to exploit the advantages of acute genetic perturbations along with pharmacological tools and conventional chronic knockout models. To prepare acute and organotypic slices, we use the latest Leica 1200S Vibratome. Our setup was built on the I-shaped Nikon FN1 upright microscope, and is positioned on a Supertech anti-vibration table. We use a water-immersion 40x CFI Apo objective with NA 0.8 and a working distance of 3.5 mm. Infrared DIC is utilized to enhance contrast in our samples and patching of selected neurons is aided by a Hamamatsu near infrared ½-inch CCD camera (C7500-51). An epifluorescence attachment also allows visualization of cells with genetically-encoded fluorescent markers, which help to identify and target specific cell types required for certain projects. Patch-clamp electrodes are pulled by a Sutter P-1000 horizontal pipette puller. Two Luigs & Newmann micromanipulator systems help us to carry out paired recordings or extracellular stimulation combined with single cell recordings. Whole-cell recordings are made in voltage- or current-clamp mode using an Axoclamp 2B amplifier from Axon Instruments. A second, almost identical setup is also available to us at the Nikon Microscopy Center located within the institute. This setup has a Nikon-C1 confocal attachment and we plan to monitor presynaptic calcium transients and postsynaptic responses simultaneously in our preparations. With the help of these electrophysiological setups, we aim to contribute to the understanding of how distinct endocannabinoid-related signaling pathways regulate certain forms of synaptic plasticity.